趋势图+热图+基因标注+GO,Pathway功能注释
简介
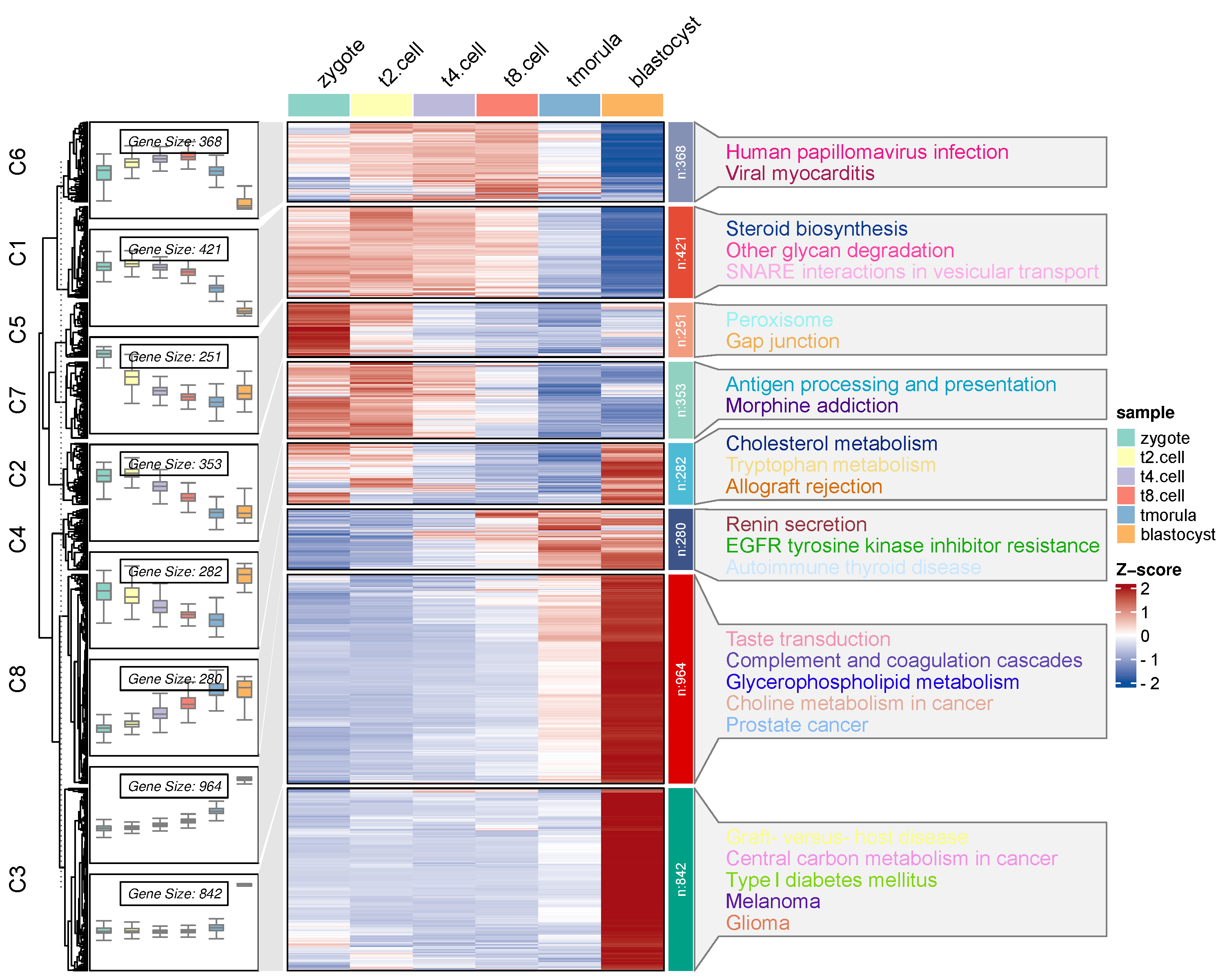
使用mfuzz或者kmeans算法将时间序列转录组数据分成不同的cluster,然后根据每个cluster的基因进行GO和Pathway等分析,获得每个cluster的注释信息(不能太多,挑选topN),然后绘制每个cluster的线图(或者box图)+热图+基因+功能注释。基因和功能注释为可选项。调用ClusterGVis包
数据说明
输入数据为矩阵形式,第一列是基因名(唯一),第2+列是表达值(一般为标准化的fpkm,tpm等,log2转换)。若有输入每个cluster的注释信息,则输出包括注释信息。第一列必须是C1,C2等。输出包括:1)elbow图,可以根据拐点确定合适的cluster数目;2)线图+热图+功能注释组合图,或者box+热图+功能注释组合图;3)每个基因所在的cluster列表
论文例子
Functional Genome Analyses Reveal the Molecular Basis of Oil Accumulation in Developing Seeds of Castor Beans. fig 1b
如何引用?
建议直接写网址。助力10000+篇
(google学术),9500+篇
(知网)论文
正式引用:Tang D, Chen M, Huang X, Zhang G, Zeng L, Zhang G, Wu S, Wang Y.
SRplot: A free online platform for data visualization and graphing. PLoS One. 2023 Nov 9;18(11):e0294236. doi: 10.1371/journal.pone.0294236. PMID: 37943830.
方法章节:Heatmap was plotted by https://www.bioinformatics.com.cn (last accessed on May 4, 2026), an online platform for data analysis and visualization.
致谢章节:We thank Mingjie Chen (Shanghai NewCore Biotechnology Co., Ltd.) for providing data analysis and visualization support.